The goal of this tutorial is to demonstrate how to run phastCons and phyloP using the phastWeb interface.
The first step is to upload a sequence alignment file.
Maximum File size: 40 MB
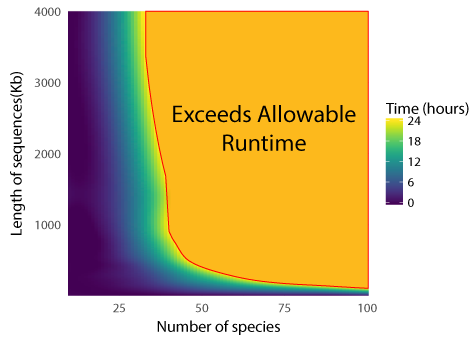
Run times are estimated based on the size of the alignment and the number of species. Your job will be rejected if the estimated run time exceeds 24 hrs. By example, an alignment with 20 species and a length of 115kb has a similar run time of 10 minutes to an alignment with 75 number of species and a length of 2kb. For estimated run times, please refer to this graph.
The reference sequence should be the first one in the alignment.
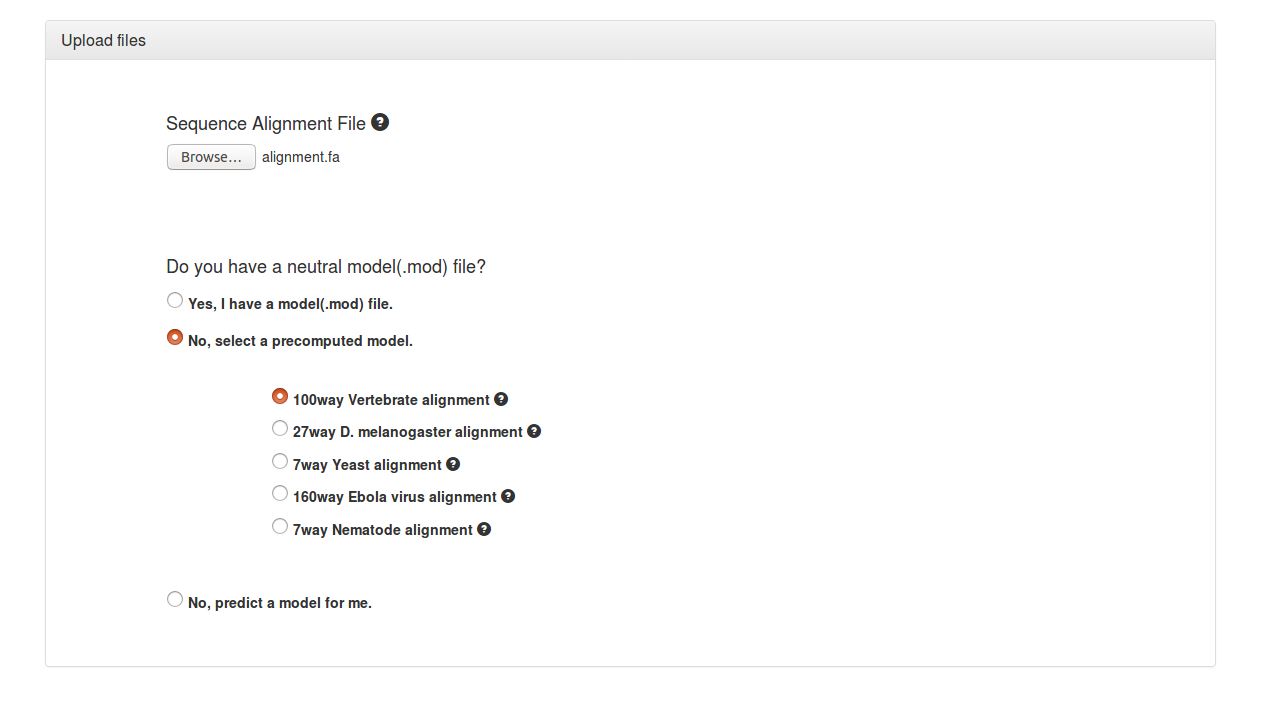
It is usually best to either choose from the options of precomputed models (Vertebrate, D. melanogaster, Yeast, Ebola virus, Nematode) available on phastWeb or to upload a pre-computed model of neutral evolution (e.g., estimated from fourfold degenerate synonymous sites or ancestral repeats) if one is available from a previous PHAST analysis. Most users, however, will not have such a model and will have to estimate one from the alignment file.
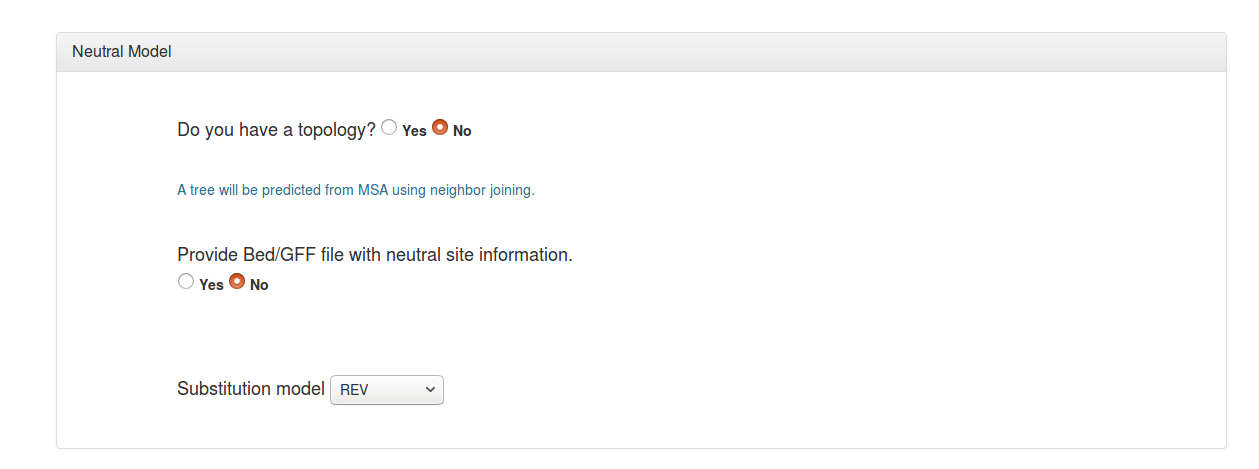
A phylogenetic tree topology is needed to estimate the neutral model. The user can specify a tree topology in “New Hampshire” or "Newick” format (*.nh), if one is known. Alternatively, a tree topology can be estimated from the alignment file data using the neighbor-joining method (neighbor from PHYLIP is used).
Maximum File size: 20 MB
If you have annotations that allow you to identify a subset of sites in your alignment that are likely to be free from selection (such as ancestral repeats or intergenic regions), you can specify these sites using a “feature” file (in GFF or BED format). The coordinates in this feature file must refer to the first (reference) sequence in the alignment. If you do not specify such a file, a model will be estimated from all sites in your alignment (this is generally okay for genomes in which most sites evolve neutrally).
You will have the option to select a nucleotide substitution model for the neutral model. The default general reversible (REV) model is a good general-purpose choice, but other commonly used substitution models are also available.
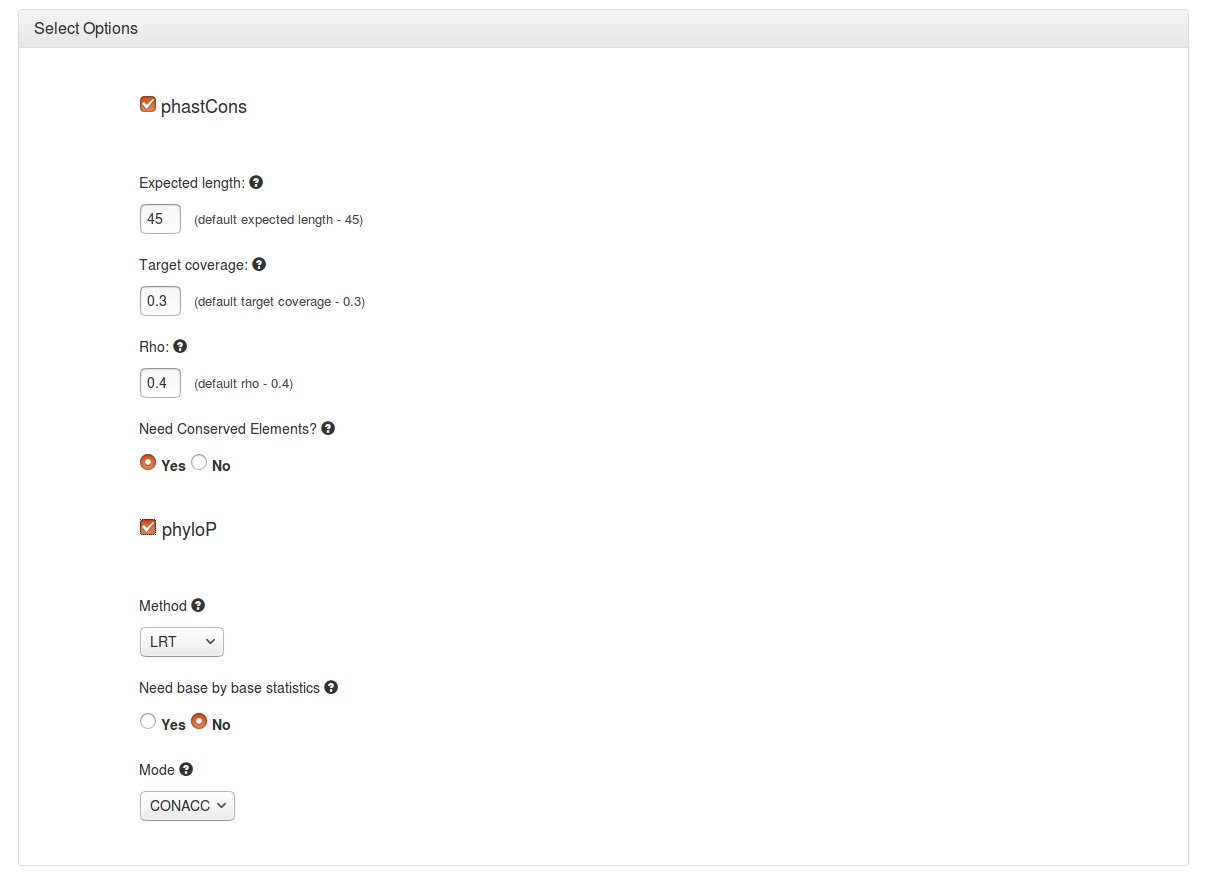
Users have the option to run either phastCons or phyloP, or both programs together.
phastCons options
▪ Expected length - Sets transition probabilities such that the expected length of a conserved element is as specified. The default of 45 is used in the example.
▪ Target Coverage - Constrain transition parameters such that the expected fraction of sites in conserved elements is as specified. The default of 0.3 is used in the example.
▪ Rho - Sets the scale (overall evolutionary rate) of the model for the conserved state to be rho times that of the model for the non-conserved state (which is defined by the neutral model. The default of 0.4 is used in the example.
▪ Conserved elements - Predicts discrete elements using the Viterbi algorithm.
phyloP options
▪ Method - Used to compute p-values or conservation/acceleration scores. A number of methods are available.
▪ Need base by base statistics - Compute statistics per site, and then compute site-specific conservation (acceleration) scores, in a method-dependent way. With 'SPH', output includes mean and variance of posterior distribution, with LRT and SCORE it includes the estimated scale factor(s) and test statistics.
▪ Mode - Used to compute one-sided or two-sided p-values to indicate conservation, acceleration or an unexpected departure from neutrality. A number of modes are available.
The user has to provide an email address before submitting the job to be notified when the results are ready.
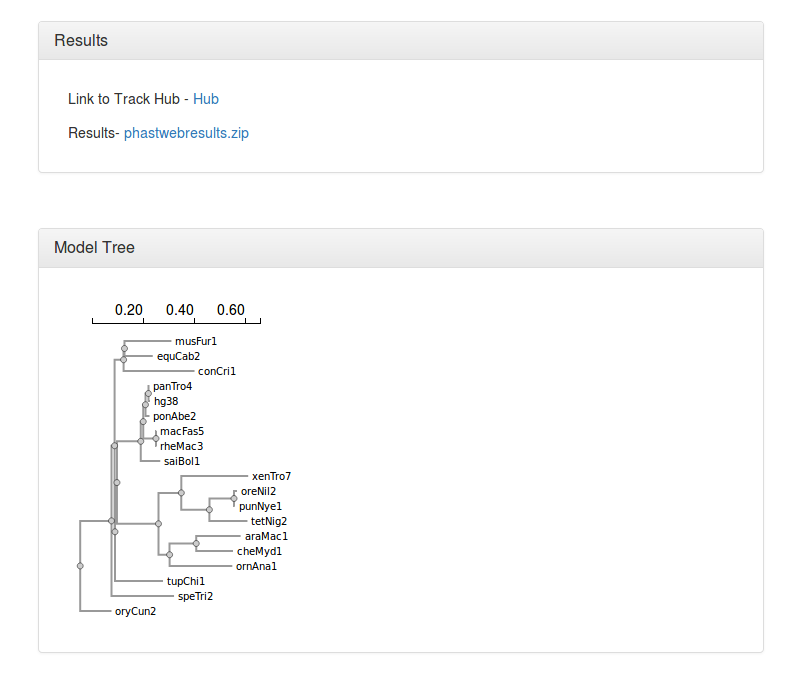
The results include three main parts
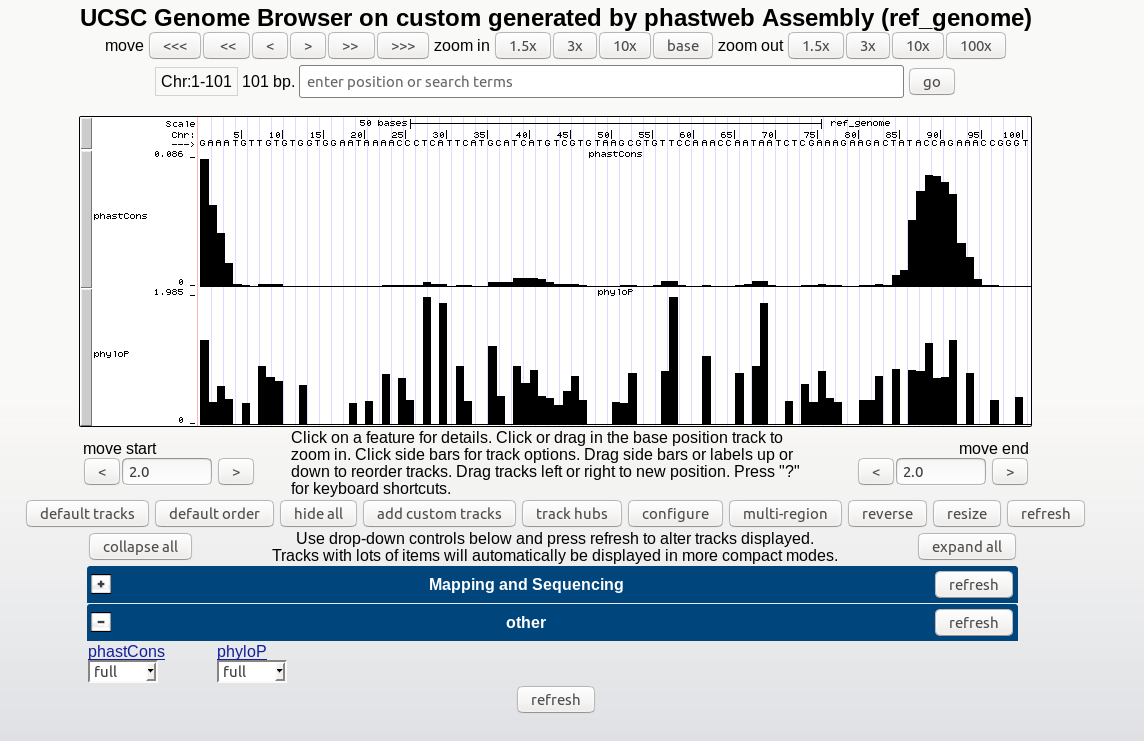
▪ A link to the UCSC Genome Browser's track hub displaying the generated conservation scores.
▪ A zip file containing the phastCons and/or phyloP results (wig files), the tree topology (if estimated by neighbor), the neutral phylogenetic model estimated by phyloFit, and a directory (myHub) which includes the bigwigs as well as the other configuration files requred for viewing the tracks in the UCSC Genome Browser.
▪ An image displaying the neutral phylogeny used for the analysis.
Last modified: February 18 2026