|
Drosophila Polymorphism and Divergence Data
|
|
This website contains information on the Drosophila melanogaster polymorphism and divergence data used
by INSIGHT. More details are available in the Supplementary Materials of
(Gronau et al., Mol Biol Evol, 2013).
More information on INSIGHT can be found here. An application of INSIGHT within D. melanogaster miRNAs was demonstrated within this additional article: (Mohammed et al., in review for RNA 2014). |
Contents
1. Polymorphism data
2. Outgroup sequence data and ancestral priors
3. Filters
4. Putative neutral sites
5. Genomic blocks
|
|
1. Polymorphism data
|
|
Drosophila melanogaster polymorphism sequence data was obtained from the Drosophila Genetic Refrence Panel (DGRP). This resource contains the complete genome sequences of
205 unrelated fly lines from a homogeneous Raleigh, North Carolina fruit-fly population.
Genotype calls for these lines were extracted from the
Variant Call Format (VCF) files downloaded from the DGRP freeze 2 website on May 2013. We considered only single nucleotide polymorphisms (SNPs) and discard sites deemed as structural variants within these VCF files. All other positions not reported in the VCF files were assumed to be monomorphic for the reference
allele (according to UCSC dm3 / BDGP Release 5).
The polymorphism data were summarized by recording for each position in dm3
the allele count for each of the four basses (A,C,G, and T) across the 205x2=410
chromosomes. Sites with more than two observed alleles (i.e. tri-allelic sites, etc) were masked. Additionally, sites with missing data for 5 or more of the 205 fly lines were masked (see filters).
|

205 Drosophila melanogaster lines from a single Raleigh, Nord Carolina populations whose genomes have been
sequenced to high coverage by the Drosophila Genetic Reference Panel.
Taken from the DGRP online resource.
|
|
|
2. Outgroup sequence data and ancestral priors
|
|
Divergence was inferred using four melanogaster-subgroup outgroup genomes:
D. simulans (droSim2), D. sechellia (droSec2), D. yakuba (droYak3) and D. erecta (droEre2). We created a custom 5-way alignment of these outgroup species and D. melanogaster (dm3) using the LASTZ and chain/net procedure prescribed by the UCSC genome browser. All genome assemblies, excluding D. simulans, were downloaded from the UCSC genome browser. We utilized a higher-quality, recently-released D. simulans genome assembly, named droSim2, in our custom alignment.
For each position in dm3, we recorded the aligned base
from each of the 4 non-melanogaster fruit-fly species, or an indication that no syntenic alignment
was available at that position (see filters).
A prior distribution for the ancestral state (Z) was computed
for all non-filtered sites in dm3, by assuming
a phylogeny estimated from four-fold degenerate sites (see Figure), and
applying the postprob.msa function in
RPHAST.
The D. melanogaster (dm3) sequence was masked in this computation, so that the computed
distribution corresponds to the distribution over the bases in the ancestral sequence
(Z) given the other four genomes,
which is used as a prior distribution (P(Z|O)) by the INSIGHT model.
|
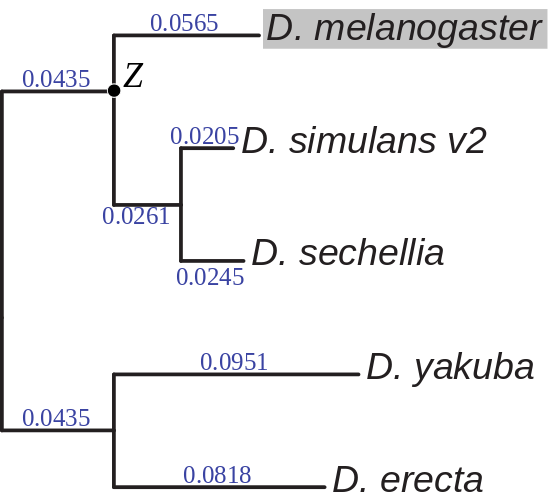
The phylogeny assumed when estimating divergence rates (λ) and prior probabilities
for the ancestral states (Zi). Branch lengths are given in expected number
of differences between haploid chromosomes per base. The phylogeny was inferred using four-fold degenerate
sites within D. melanogaster protein coding genes (FlyBase r 5.46) using the phyloFit utility from the PHAST package.
|
|
|
3. Filters
|
|
DGRP freeze 2 datasets were only provide for the homozygous blocks of the autosomes and the X chromosomes (i.e. 2L, 2R, 3L, 3R, 4, X). Polymorphic sites within heterozygous blocks (i.e. 2LHet, 2RHet, 3LHet, 3RHet, XHet, and YHet) or for the mitochondrial DNA were not provided. Thus, our analysis was restricted to the homogous chromsomal regions. We applied various filters to reduce the impact of technical errors from alignment, sequencing,
genotype inference, and genome assembly. Our filters included repetitive
sequences (simple repeats), recent transposable elements, recent segmental duplications, and CpG site pairs. CpG site pairs (prone to hypermutability) were identified as position
pairs having a “CG” dinucleotide in the D. melanogaster reference genome. Non-syntenic regions, as identified from synthenic blocks residing on low-quality Nets, and gaps in the outgroup alignment were hard-masked (by “N”s) individually in each
outgroup genome. This uncertainty was incorporated when estimating the prior distribution over
the ancestral sequence (Z, see above).
Sites with missing data in greater than or equal to 5 of the 205 fly lines were masked
out completely (see above). Additionally, sites with more than two observed
alleles in the fly population data (i.e. tri- or quad-allelic sites) were masked. We treated lines without a reported genotype and lines with "N" base assignments as lines with "missing" data. At polymorphic sites where less than 5 individuals contained missing data, we subsampled 200 fly lines without replacement in order to arrive an a uniform number of 400 alleles per site. This subsampling approach is a common method utilized in processing genotype data with missing information, and offers consistency and accuracy in the treatment of polymorphisms within INSIGHT. |
Filters used for INSIGHT analysis of D. melanogaster data.
Links provided to BED files and genomic coverage is given in megabases.
|
|
|
4. Putative neutral sites
|
|
Estimates of neutral model parameters were computed by considering a collection of putative neutral sites that pass our
filters. The collection of putative neutral sites was determined
by eliminating sites likely to be under selection: (1) exons of annotated protein-coding genes and the 50
bp flanking them, and (2) conserved noncoding elements
(identified by phastCons) and 25 bp flanking them. While a fraction of the remaining sites is likely to be functional, this set should be dominated
by sequence evolving under neutral drift. |
|
|
5. Genomic blocks
|
|
For estimation of genome-wide neutral polymorphism and divergence rates, we used a
fixed collection of 1kb non-overlapping windows.
We used the putative neutral sites in each 1 kb window to estimate a neutral polymorphism rate (θb)
and a neutral divergence rate (λb), and those estimates were then associated with the appropriate
genomic block.
|
|
|