fitCons, the fitness consequences of functional annotation, integrates functional assays (such as ChIP-Seq) with selective pressure inferred using the INSIGHT method. The result is a score ρ in the range [0.0-1.0] that indicates the fraction of genomic positions evincing a particular pattern (or "fingerprint") of functional assay results, that are under selective pressure. As these scores show the selective pressure consequences of patterns of functional genomic assays, they can vary per cell-type just as functional assays do. Scores combine conservative and adaptive selective pressures and may be used an a relative indicator of the potential for interesting genomic function, with higher scores indicating more potential.
These tracks show the genome-wide scores for fitCons, as well as the particular pattern of functional assays (covariates) associated with each genomic position in each of three ENCODE cell lines: HUVEC, H1-hESC (WA01), and GM12878. In addition, a fourth set of scores and covariates (i6) represents scores integrated across cell-types, and is likely to be the most useful for researchers investigating cell-types that are different from the three types provided here.
Pre-publication release of fitCons scores. First made available 5-Jul-2014, last updated 23-Jul-2014.
Calculating the FitCons Score
Covariate Selection
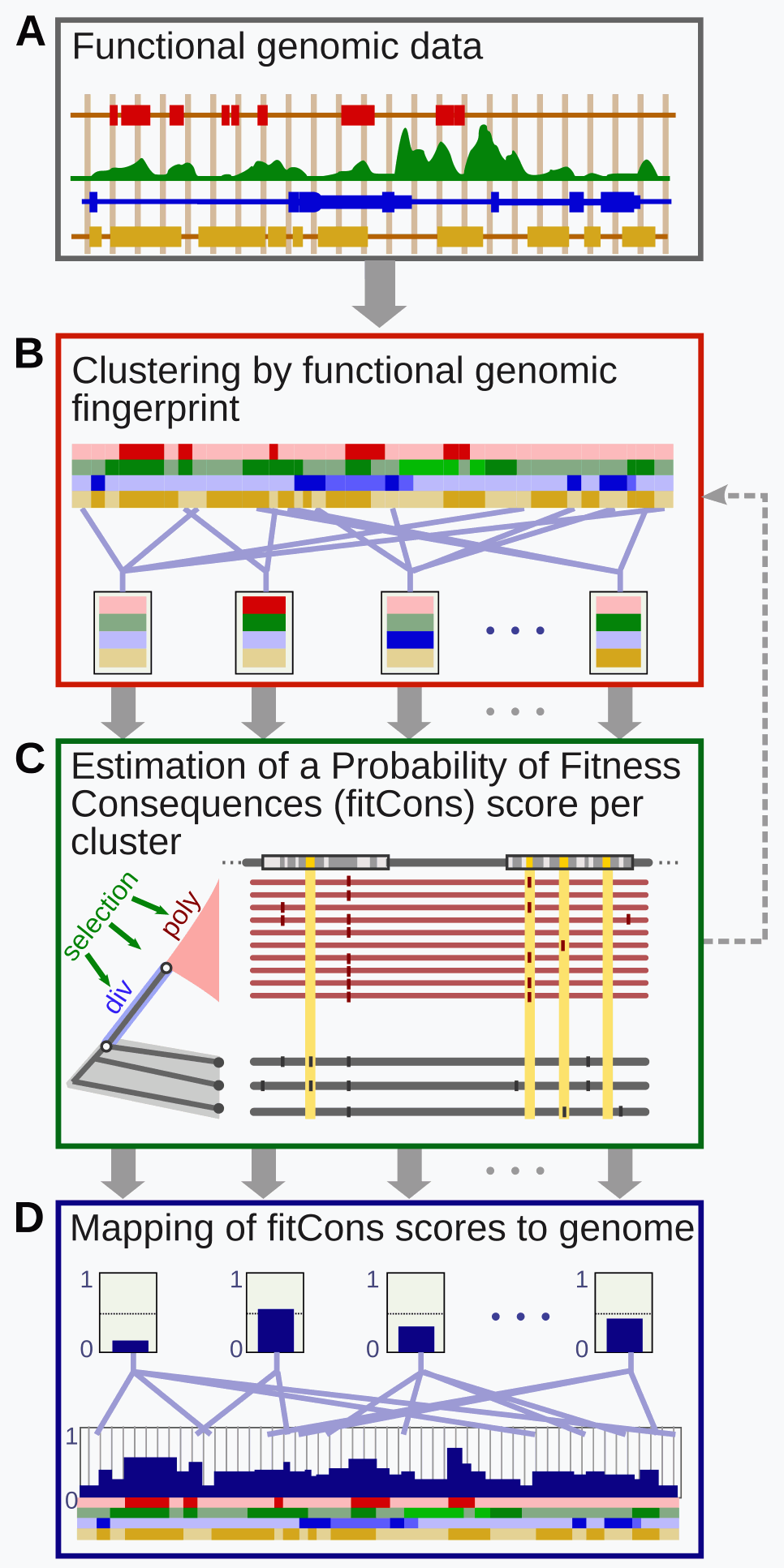
Four covariates (A) are obtained for each cell type: DNase I peaks, Normalized RNA-Seq Read Depth (RPM), Chromatin State (ChromHMM class) and GENCODE annotation as protein coding CDS. The last of these is common to all cell types. Each data set is then quantized into as small number of classes (B), assigning each position in the genome, one class, from each data set.
- DNase I HS Peaks: 2-Narrow Peak, 1-Broad Peak (and not Narrow Peak), and 0-No Peak
- RNA-Seq Read Depth: 3-High Depth, 2-Medium Depth, 1-Low Depth, and 0-No Signal
- CDS Annotation: 1-Annotated as CDS, or 0-Not Annotated
- Chromatin State: [1-25] Chrom HMM class assigned to that position, or 26-No Class available.
This generates a total of 3x4x2x26=624 unique "fingerprints" with each genomic position evincing exactly one finger print in each cell-type (B). All positions associated with a particular fingerprint are grouped together into a common functional class.
Evolutionary Genomic Data
Selective pressure for each functional class is inferred (C) from the distribution of human polymorphism and primate divergence, relative to nearby neutrally evolving loci. Human polymorphism data is drawn from position-wise variation among 54 unrelated human individuals from the 69 sequences released by Complete Genomics. Divergence data is derived from an the most recent common ancestor of Human and Chimpanzee, which is inferred using Chimpanzee (panTro2), Orangutan (ponAbe2), and rhesus Macaque (rheMac2) reference genomes. Putative neutral loci are identified by removing a window around known conserved and protein coding genomic positions, as well as a number of technically undesirable genomic positions (such as unmappable regions). See INSIGHT paper references below for more details on these data sets.
Applying INSIGHT
INSIGHT was applied (C) to infer a maximum likelihood estimate of the fraction of genomic positions under selective pressure (ρ) for each of the 624 functional classes. This calculation was performed separately for each cell type. The fitCons score associated with a functional class, was then assigned to all genomic positions in that class (D). A curvature based method was employed to identify the standard error in for each estimate and only classes with a standard error of less than .4xρ were considered in the published analysis. Positions with higher uncertainty are provided in this track, using different colors to indicate level of confidence. While additional INSIGHT model estimated were generated (EA and EW), they were not used in the analysis and are not provided here.
Integrating Scores (i6)
To integrate scores across cell types, a two phase method was used. First all positions demonstrating a fingerprint in any cell type were added to a collective class for that fingerprint, then INSIGHT was run to generate a fitCons score for that fingerprint. This produced a uniform ranking of fingerprints by aggregate fitCons scores, with a caveat that positions might be included in more than one functional class. In the second phase, each genomic position was assigned to the fingerprint that scored the highest from among the three finger print generated by the three cell types (using the phase one scoring). INSIGHT was run a second time on each new collection of positions to generate the final fitCons scores for this partition of the genome. In the second phase, each genomic position was included in exactly one functional class. Tracks provided for this integrated include fitCons score, original cell type source for each genomic position, and covariates for the position, in the source cell type.
Tracks
Due to technical genome browser limitations, the 4 classes (3 cell types plus integrated data) are represented as 9 separate tracks, which should generally be viewed in pairs. Each pair should reflect the covariates and the scores generated via those covariates. The four classes are designated:
- HU (HUVEC),
- H1 (H1-hESC),
- GM (GM12878), and
- I6 (Multicell).
Each class contains a:
- {Class}-Scr (score) trackset providing the fitCons score for that class, and a
- {Class}-Cov (covariates) trackset showing the quantized covariates that define the genomic finger print at each genomic position.
The additional track is the HU-ScrD track, which provides the divergence-only based measure of fitCons mentioned in our paper (References: below). The ScrD track is available only for the HUVEC cell line. In addition, the I6-Cov track set contains a subtrack labelled "Cell Line" that indicates the cell type that served as the source for that each position's covariates.
Display Conventions
For genome wide viewing, fitCons scores are best viewed within a range 0.0 to 0.80 (the default). When focusing on non-coding regions, the range .05 to .35 may be most appealing as nearly all non-coding classes have scores in this range, while nearly all coding classes have scores > .40.
Covariate Display Colors
| Color | Covariate | Description |
| | CDS | GENCODE Annotated Exonic Protein Coding CDS |
| | RNA-Seq | Group 3 - Greatest read depth |
| | RNA-Seq | Group 2 - Medium read depth |
| | RNA-Seq | Group 1 - Lowest non-zero read depth |
| | DNase-I HS | Narrow Peak (high signal) |
| | DNase-I HS | Broad Peak, but not narrow peak (low signal) |
|
For standard ChromHMM classes
click here (opens new window).
|
Cell Line Source -
Multicell Covariate Track
| Color | Description |
| | (H1) H1-hESC (WA01) |
| | (HU) HUVEC |
| | (GM) GM12878 |
A curvature based estimate of the maximum likelihood estimate for INSIGHT ρ was generated and used to estimate a simple single-tailed p-value for rejecting the conservative null hypothesis that ρ<=0. These p-values determine the color of the score display at each genomic position. All positions in a functional class have the same score and confidence value.
fitCons score colors and statistical uncertainty.
| Color | Description |
| | High confidence values used in paper (p<~.003) |
| | Likely Significant (p<.05) |
| | Likely Informative (p<.25) |
| | Best estimate (p>=.25) |
Downloads
Genome wide fitCons scores are available here.
Cell scores and covariates are organized into directories as:
- hu - HUVEC
- h1 - H1-hESC
- gm - GM12878
- i6 - Scores integrated across cell types.
If you are uncertain which score set to use, the i6 might be the best starting place followed by the cell-type most like your cell-type of interest.
Within each of these directories are two subdirectories:
- scores - fitCons scores, grouped according to statistical significance.
- cov - quantized covariates used to define the fingerprints that group genomic positions into disjoint classes.
Both of these subdirectories contain the data used to define the browser tracks visible on our browser mirror.
Covariate data is in the cov subdirectories in bigBed format.
Within the scores subdirectory there are 4 files providing scores for disjoint regions, in bigWig format:
- fc-XX-0.bw - highly significant scores (approx. p<.003), the only ones used in out paper and displayed as dark blue on our browser track. Use these scores, and these scores only, for comparison to our reported results.
- fc-XX-1.bw - significant scores (approx. p<.05).
- fc-XX-2.bw - informative scores (approx. p<.25).
- fc-XX-3.bw - other scores (approx. p>=.25).
While all scores represent the maximum likelihood estimate for the fraction of sites under selective
pressure, estimates from less significant classes fail simple one-tailed p tests for rejection of the
null hypothesis that fraction of sites under selection is zero. However, these scores are provided for display
purposes as they may still represent informative biases.
Versions
- V1.00 - Initial release, Jul-2014
- V1.01 - Bug Fix, 28-Aug-2014. Corrected a bug that caused all genomic positions in annotated CDS, with no RNA-Seq signal (RNASeq class
0, of 0,1,2,3) to be grouped into the corresponding class that was NOT annotated as a CDS, but had a High RNA Seq signal (3, of 0-3).
References
Gulko B, Gronau I, Hubisz MJ, Siepel A. 2014.
Probabilities of Fitness Consequences for Point Mutations Across the Human Genome
.
doi: http://dx.doi.org/10.1101/006825
Gronau I, Arbiza L, Mohammed J, Siepel A.
Inference of Natural Selection from Interspersed Genomic Elements Based on Polymorphism and Divergence.
Mol Biol Evol, 30(5):1159-1171, 2013.
doi: 10.1093/molbev/mst019
Arbiza L, Gronau I, Aksoy BA, Hubisz MJ, Gulko B, Keinan A, Siepel A. 2013.
Genome-wide inference of natural selection on human transcription factor binding sites
.
Nat. Genet. doi: 10.1038/ng.2658